Automation of the analysis
If you want to study many atomic resolution images of the same type of structure, you can save time and effort by using tools in Atomap for automation, by setting process parameters.
atomap.process_parameters is a module offering classes of predefined process parameters for some types of structures:
Perovskite Oxides (projected along the [110] direction), for all sublattices (A, B and Oxygen)
A Generic structure
This tutorial will first show how the use of these process parameters to make the procedure for finding the full atom lattice more automatic. Currently, predefined process parameters are available for a limited number of materials and projections. The last part of this tutorial aims to show how such parameters can be made for new materials.
Finding atom lattices with process parameters
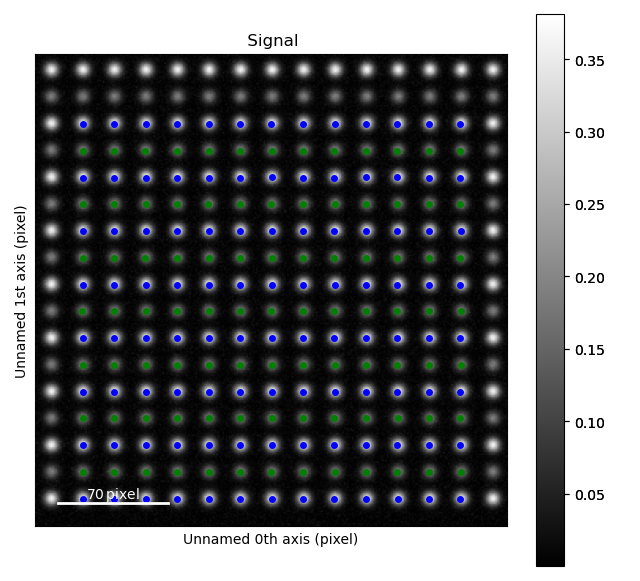
In this tutorial we will use the predefined process parameter PerovskiteOxide110 PerovskiteOxide110, and a dummy image designed to look like an HAADF image of the perovskite SrTiO3.
It contains various parameters and names for processing a perovskite oxide structure projected along the [110] direction.
The main function atomap.main.make_atom_lattice_from_image() takes the atomic resolution signal, process parameters and optimal feature separation.
This means that you probably need to run atomap.atom_finding_refining.get_feature_separation() and find the best pixel separation first.
>>> import atomap.api as am
>>> s = am.dummy_data.get_two_sublattice_signal()
>>> process_parameter = am.process_parameters.PerovskiteOxide110()
>>> atom_lattice = am.make_atom_lattice_from_image(s, process_parameter=process_parameter, pixel_separation=14)
1/2
2/2
1/2
2/2
Depending on the size of the dataset, this can take a while. The processing will:
Locate the most intense atomic columns (A-cations, Strontium).
Refine the position using center of mass.
Refine the position using 2-D Gaussian distributions
Find the translation symmetry using nearest neighbor statistics, and construct atomic planes using this symmetry.
Locate the second most intense atomic columns (B-cation, Titanium), using the parameters defined in the model parameters
“Subtract” the intensity of the A-cations from the HAADF image
Refine the position of the B-cations using center of mass
Refine the position of the B-cations using 2-D Gaussian distributions
Construct atomic planes in the same way as for the first sublattice.
This returns the atom_lattice object, which contains the sublattices of both A and B cations.
>>> atom_lattice.plot()
Making process parameters
You can make customized process parameters for the structures you are studying in order to make analysis with Atomap more automated. In the atomap.process_parameters you can add your own classes with process parameters for your own structures. This section will give an example of how to construct these classes. It is assumed you have read Become a contributor first, where there is some information on how to develop code for Atomap.
YourStructure
Our example is the imaginary YourStructure, which consists of two sublattices, SublatticeA and SublatticeO.
SublatticeA is a sublattice consisting of heavy transition metal atoms, while SublatticeO is oxygen (light atoms).
This means that SublatticeA will have good contrast in ADF-images, while SublatticeO is only visible in ABF-images (together with A, of course).
The example is constructed to give an insight in how you program the parameters to use the different images to find the atom positions.
Of course, if all the sublattices in your real material are best resolved in ADF, this image can be used for all.
First, you should create a class for each of the sublattices in the structure.
The sublattice process parameter class contains information about how the sublattice should be refined, zone axes and which order it has in the structure.
The most intense sublattice has order 0, the second most intense sublattice has order 1, etc.
Also, the sublattice inherits the class SublatticeParameterBase.
As the heavy A-atoms are best resolved in ADF/HAADF images, the dark field image will be used to find the atom positions of A.
A class for the process parameters for SublatticeA can look like this:
class SublatticeA(SublatticeParameterBase):
"""Docstring describing your sublattice.
"""
def __init__(self):
SublatticeParameterBase.__init__(self)
self.color = 'red'
self.image_type = 0
self.name = "A"
self.sublattice_order = 0
self.zone_axis_list = [
{'number': 0, 'name': '100'},
{'number': 1, 'name': '111'},
]
self.refinement_config = {
'config': [
['image_data_modified', 1, 'center_of_mass'],
['image_data', 1, 'center_of_mass'],
['image_data', 1, 'gaussian'],
],
'neighbor_distance': 0.35}
In this class, the color of the markers used to show atom positions in the plots will be red, and the name of the sublattice is ‘A’.
With
image_type = 0, the atomic resolution image used to find atom positions will not be inverted. In dark field images the atoms are bright, so no inversion is needed.YourStructure has two zone axes, 100 and 111. These are added in the
zone_axis_listas shown.refinement_configis the refinement configuration. In this example the positions are refined three times as follows:Atom positions are refined one time by using center-of-mass on an image where the background has been removed, noise has been filtered with PCA and the image is normalized.
Atom positions are refined one time by using center-of-mass on the original image.
Atom positions are refined one time by fitting 2D-Gaussians to the original image.
An appropriate
neighbor_distancemust be given to set the mask size for the fitting of the Gaussians. Here, it is 35 % of the distance to the nearest neighbor.
To find the atom positions in SublatticeO, an ABF image is used.
class SublatticeO(SublatticeParameterBase):
"""Docstring describing your sublattice.
"""
def __init__(self):
SublatticeParameterBase.__init__(self)
self.color = 'green'
self.image_type = 1
self.name = "O"
self.sublattice_order = 1
self.zone_axis_list = [
{'number': 0, 'name': '100'},
{'number': 1, 'name': '111'},
]
self.sublattice_position_sublattice = "A"
self.sublattice_position_zoneaxis = "111"
self.refinement_config = {
'config': [
['image_data_modified', 1, 'center_of_mass'],
['image_data', 2, 'gaussian'],
],
'neighbor_distance': 0.25}
self.atom_subtract_config = [
{
'sublattice': 'A',
'neighbor_distance': 0.35,
},
]
In this class, the color of the markers used to show atom positions in the plots will be green, and the name of the sublattice is ‘O’.
With
image_type = 1, the atomic resolution image used to find atom positions will be inverted. This is because in the bright field image the atoms are dark and surroundings are bright. For Atomap to work, the atoms must be the most intense features.The zone axes is the same as for the other sublattice, they are both a part of YourStructure.
sublattice_position_sublattice = "A"andsublattice_position_zoneaxis = "111": The O columns are located between the columns in sublattice “A” in the direction of the zone axis 111. This setting is used to find the initial positions of the atomic columns inSublatticeO.atom_subtract_configis the configuration for how the brighter sublattices should be removed from the image prior to fitting the less bright sublattices. Here, the sublattice ‘A’ is removed from the image. An appropriateneighbor_distancegives the size of the mask around the A atoms. If no atoms should be removed from the image, this list can be removed from the class (as forSublatticeAabove).refinement_configis different here, to illustrate the possibilities:Atom positions are refined one time by using center-of-mass on an image which has been inverted and with the A sublattice removed, and modified by background removal, noise filtering and normalization.
Atom positions are refined two times by fitting 2D-Gaussians to the inverted image where the A sublattice has been removed.
0.25 is found to be an appropriate
neighbor_distancefor this example structure.
Play around with the refinement configurations and neighbor distances to find what works on your images and structures.
The above sublattices belong to YourStructure.
This class inherits from ModelParametersBase can look like this:
class YourStructure(ModelParametersBase):
"""Docstring describing your sublattice
"""
def __init__(self):
ModelParametersBase.__init__(self)
self.name = "Wondermaterial"
self.peak_separation = 0.127
self.sublattice_list = [SublatticeA(), SublatticeO()]
An important setting here is the peak_separation.
The peak separation is a distance in nanometer, a little less than half the distance between the atoms in ‘A’.
The number is used to find the pixel_separation for the initial peak finding for the brightest sublattice.
Therefore, the scale of the image must be calibrated prior to processing.